Evaluating read mismatch statistics: mapping and de novo¶
Looking at read mismatch profiles by mapping to a reference¶
One of my favorite approaches to looking at error bias in read data sets is not to just use the Q score, but to do something empirical instead – for example, if you already have a reference, you can map the reads to the reference and look at mapping mismatches.
Do a mapping with Bowtie2:
cd ~/notebooks/ecoli
curl -O https://s3.amazonaws.com/public.ged.msu.edu/ecoliMG1655.fa.gz
gunzip ecoliMG1655.fa.gz
bowtie2-build ecoliMG1655.fa ecoli
gunzip -c ecoli-R1.fq.gz | bowtie2 -p 4 -x ecoli -U - -S ecoli-R1.sam
then use a custom script to look at where in the read mapping there are mismatches:
~/notebooks/sam-scan-errhist.py ecoliMG1655.fa ecoli-R1.sam -o ecoli-mapped.errhist
Looking at the read mismatch profile de novo¶
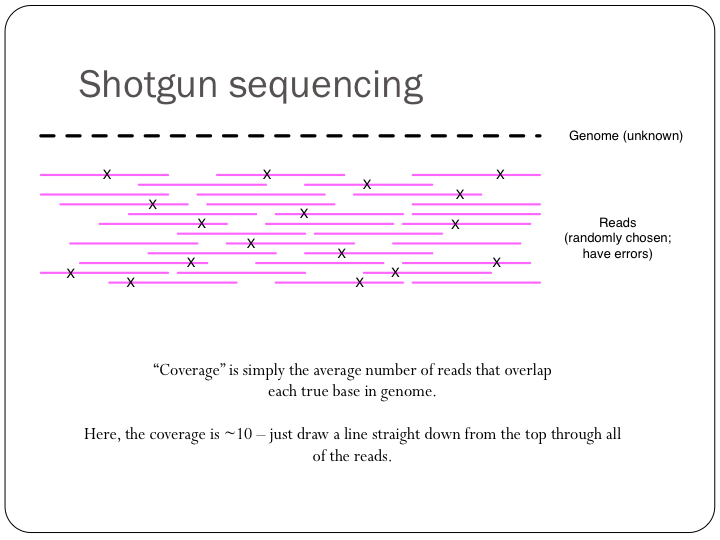
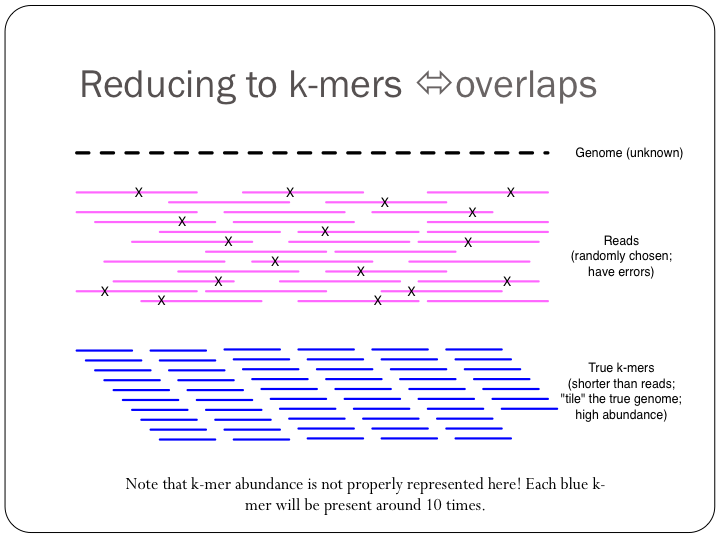
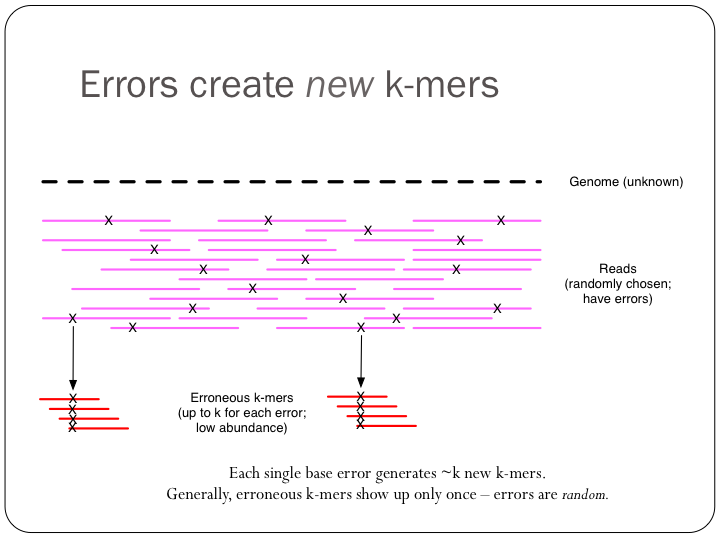
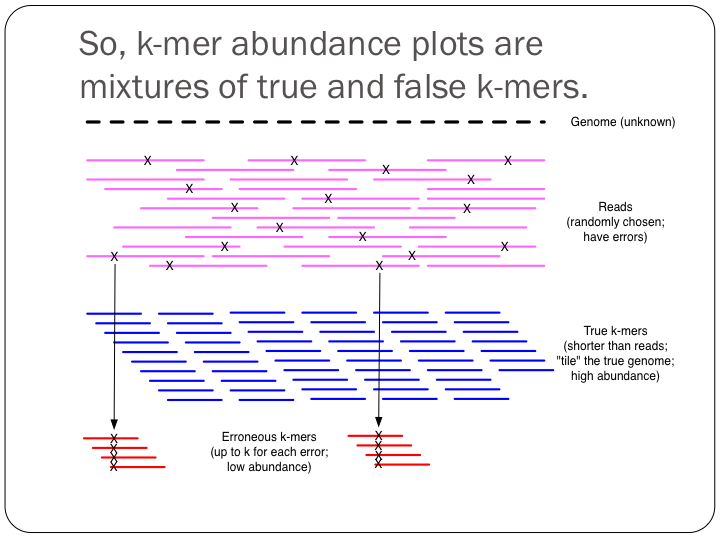
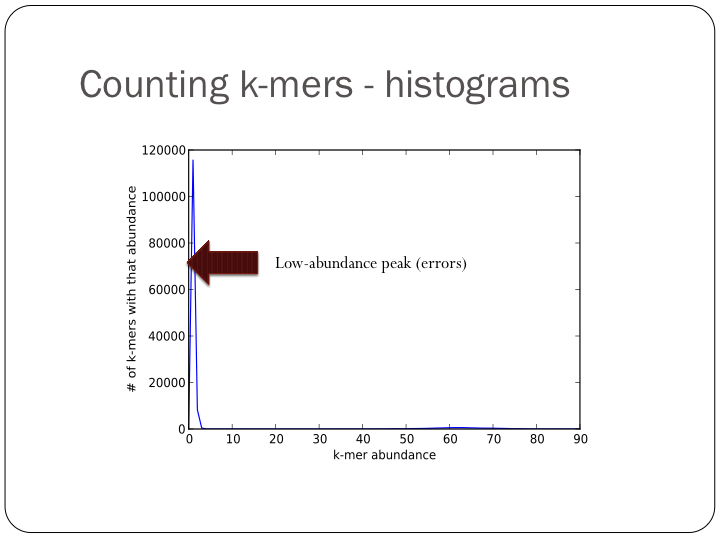
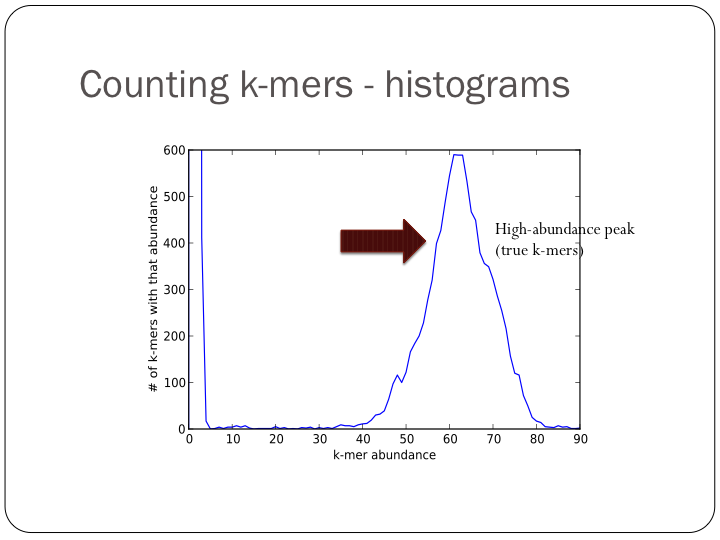
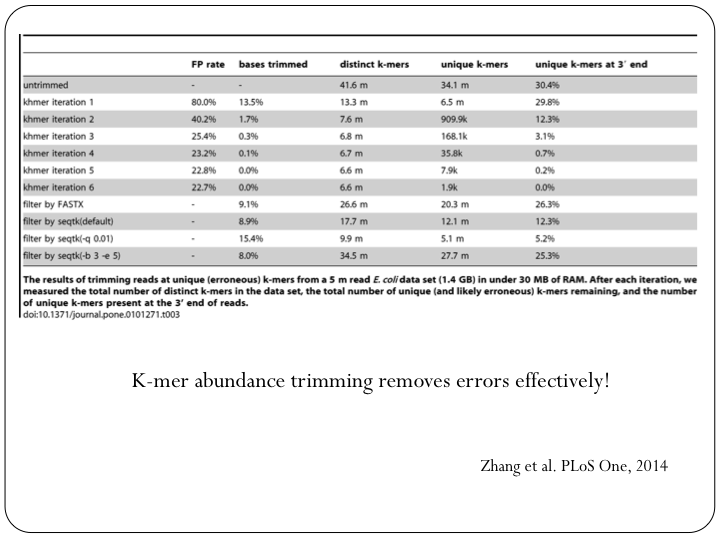
To try this out with khmer, do:
cd ~/notebooks/ecoli
load-into-counting.py -M 1e8 -k 21 ecoli.kh ecoli-R1.fq.gz
~/notebooks/report-errhist-2pass.py -C 1 ecoli.kh ecoli-R1.fq.gz > ecoli-kmer.errhist
(Note, you don’t want to do k-mer abundance trimming in general, but it’s useful for evaluation.)
Look at the errhist files using ‘less’ or ‘tail’... Or plot them.
Can we do this for RNAseq? (Use the -V option on report-errhist-2pass).
LICENSE: This documentation and all textual/graphic site content is licensed under the Creative Commons - 0 License (CC0) -- fork @ github. Presentations (PPT/PDF) and PDFs are the property of their respective owners and are under the terms indicated within the presentation.